The influence of reduced resolution quality scores on alignment and variant calling
BAM file size reduction and quality score binning
We have a large upcoming whole genome sequencing project with Illumina, and they approached us about delivering BAM files with reduced resolution base quality scores. They have a white paper describing the approach, which involves binning scores to reduce resolution. This reduces the number of scores describing the quality of a base from 40 down to 8.
The advantage of this approach is a significant reduction in file size. BAM files use BGZF compression, and the underlying gzip DEFLATE algorithm compresses based on shared text regions. Reducing the number of quality values increases shared blocks and improves compression. This reduces BAM file sizes by 25-35%: an exome BAM file reduced from 5.7Gb to 3.7Gb after quality binning.
The potential downside is that the reduction in quality resolution may impact alignment and variant calling approaches that rely on base quality scores. To assess this, I implemented quality score binning as part of the bcbio-nextgen analysis pipeline using the CRAM toolkit and ran alignment, recalibration, realignment and variant calling on:
- The original unbinned 40-resolution base quality BAM from an NA12878 exome.
- The BAM binned into 8-resolution base qualities before alignment.
- The BAM binned into 8-resolution base qualities before alignment and binned again following base quality score recalibration.
A comparison of alignment and variant calls from the three approaches indicates that binning has nearly no impact on alignment and a small impact on variant calls, primarily in low depth regions.
Alignment differences
We aligned 100bp paired end reads with Novoalign, a quality aware aligner. Comparison of mapped reads showed nearly no impact on total mapped reads. The plot below shows a generic delta of changes in mapped reads across the 22 autosomes alongside the increase in unmapped pairs. Out of 73 million total reads, the changes account for ~0.003% of the total reads. There also did not appear to be any worrisome patterns of loss for specific chromosomes. Overall, there is a minimal impact of quality score binning on the ability to align the reads.
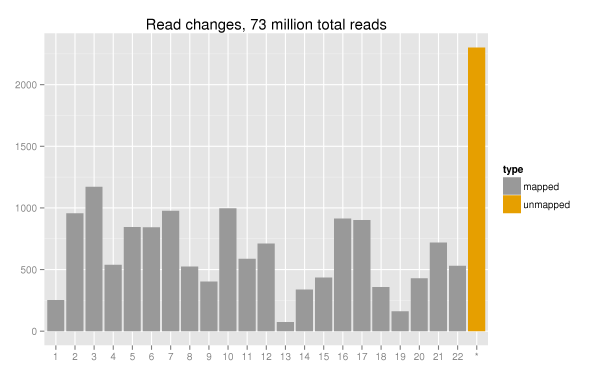
Variant call differences
We called variants using the GATK Unified Genotyper following the best practice recommendations for exomes and then compared calls from original and binned quality scores. Both approaches for binning — pre-binning, and pre-binning plus post-quality recalibration binning — showed similar levels of concordance to non-binned quality scores: 99.81 and 99.78, respectively. Since the additional binning after recalibration provides a smaller prepared BAM file for storage and has a similar impact to pre-binning only, we used this for additional analysis of discordant variants.
The table below shows the discordant differences between the 40 quality score resolution and binned, 8 quality score resolution BAMs. 40 quality discordant variants are those called with full quality score resolution but not called, or called differently, after binning to 8 quality score resolution. Conversely, the 8-quality discordants are those called uniquely after quality binning:
| Overall genotype concordance | 99.78 |
| concordant: total | 117887 |
| concordant: SNPs | 109144 |
| concordant: indels | 8743 |
| 40-quality discordant: total | 821 |
| 40-quality discordant: SNPs | 759 |
| 40-quality discordant: indels | 62 |
| 8-quality discordant: total | 1289 |
| 8-quality discordant: SNPs | 1240 |
| 8-quality discordant: indels | 49 |
| het/hom discordant | 259 |
We investigated the discordant variants further since 1.5% of the total variant calls change as a result of binning, Of the 1851 unique discordant variants, approximately half (928) fall into reproducible variants identified by looking at ensemble combinations of replicates. Of these potentially problematic discordant variants more than half are in low coverage regions with less than 10 reads:
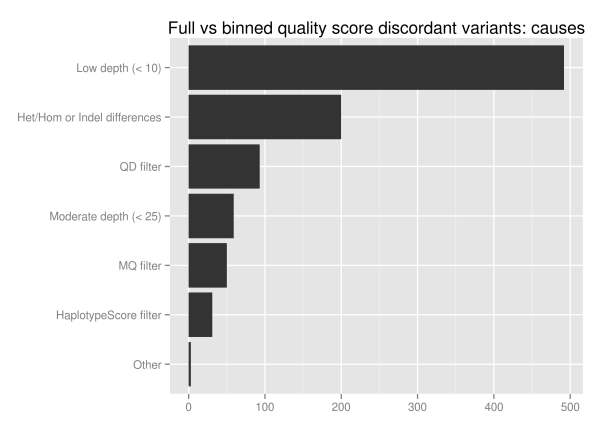
The major influence of quality score binning is resolution of variants in low coverage regions. This manifests as differences in heterozygote and homozygote calling, indel representation and filtering differences related to quality and mappability. To assess the potential impact, we looked at the loss in callable bases on a 30x whole genome sequence when moving from a minimum of 5 reads to a minimum of 10, using GATK’s CallableLoci tool. Regions with read coverage of 5 to 9 make up 4.7 million genome positions, 0.17% of the total callable bases.
| 5 read minimum | 10 read minimum | |
|---|---|---|
| Callable bases | 2,775,871,235 | 2,771,109,000 |
| Percent callable | 96.90% | 96.73% |
| Low coverage | 17,641,980 | 22,404,215 |
| No coverage/ poor mapping | 71,272,008 | 71,272,008 |
In conclusion, quality score binning provides a useful reduction in input file sizes with minimal impact on alignment. For variant calling, use additional caution in low coverage regions with less than 10 supporting reads. Given the rapid increases in read throughput that are driving the need for file size reduction, quality score binning is a worthwhile tradeoff for high-coverage recalling work.



The reduced quality range idea is very sensible, but for compatibility with CRAM I would want to use one of the same binning schemes they use (ideally a common default). That would seem sensible to avoid data loss in BAM CRAM by two different quality binning regimes being applied.
Peter Cock (@pjacock)
February 13, 2013 at 5:55 am
Peter;
Agreed. CRAMTools 1.0 does use the Illumina binning scheme as well:
https://github.com/vadimzalunin/crammer/blob/master/src/main/java/net/sf/cram/lossy/QualityScorePreservation.java#L151
It looks like CRAMTools previously had another binning scheme (NCBI binning scheme):
https://github.com/vadimzalunin/crammer/blob/master/src/main/java/net/sf/cram/lossy/Binning.java
but that no longer appears to be used, probably for compatibility. Here’s the code used to go BAM -> binned CRAM -> BAM that we’re using:
https://github.com/chapmanb/bcbio-nextgen/blob/master/bcbio/bam/cram.py#L34
Thanks for emphasizing this point.
Brad Chapman
February 13, 2013 at 7:15 am
Thanks Brad for your excellent analysis. Would you be able to provide a brief update on this during todays call? Great work!
Anonymous
February 13, 2013 at 8:55 am
Nice analysis Brad. I think within a few years we will see basecall quality scores disappear altogether.
Jeremy Leipzig
February 14, 2013 at 3:25 pm
I agree with the Jeremy – in my experience most false positive variant calls come from misaligned reads, not multiple miscalled bases happening to align to the same position. Any word on a 2 quality bin or 1 quality bin analysis?
Brendan O'Fallon
February 14, 2013 at 4:45 pm
Practically most variant callers currently make use of quality scores, especially for selecting a minimum base quality to include reads at a position. I do think the quality scores are useful for identifying sequencing artifacts but agree it’s an open question about the right quality resolution. I’m also agreed about the importance of assessing mapping quality in reducing false positives. In my experience both are useful as you work to refine and filter calls.
Brad Chapman
February 15, 2013 at 3:03 am
I just tried this with a NA12878 dataset using just 4 bins, and .bams were about 50% smaller. Variants got shuffled around a little – concordance was near 98.5% – but the unique variants identified had similar Ti/Tv suggesting that neither identified significantly more false positives. I’m also willing to gamble that base quality score recalibration will have little effect here, although I haven’t checked that yet.
Brendan O'Fallon
February 15, 2013 at 6:33 pm
Brendan;
Thanks for investigating this. The compression size is nice but the concordance difference is a bit worrisome. I’d imagine the biggest change would be how you bin the lower quality scores. GATK has quality cutoffs of 17 for SNPs and 20 for indels for excluding low quality reads. How you placed quality scores in the 4 categories could exclude or include a larger percentage of positions than with 40-category resolution. Illumina appears to have been careful about their binning strategies to consider these type of cutoffs, which helps in maintaining similar calls. Thanks again for digging more into this.
Brad Chapman
February 16, 2013 at 12:00 pm
Hi, anyone has some experience using HDF5 or the BioHDF strategy to compress SAM alignments. That compression does not require binning the quality values and therefore may not impact on genotype quality. There seems to have been some work on this compression method but did not become popular. I wonder if people have tested it and found it not useful for some reason they may want to share … :)
BW
Inti Pedroso (@intipedroso)
February 26, 2013 at 4:59 pm
Inti;
The issue is more practical than technical. While there may be advantages to alternative compression approaches (HDF5, Goby, Cram), most existing tools work with BAM format. Using a different storage file format would require converting to/from BAM during analysis, or rewriting software to deal with the format. This tends to be prohibitive for most researchers.
Brad Chapman
February 27, 2013 at 3:24 pm
Brad,
Do these results suggest that the Base Quality Score Recalibration (BQSR) step in GATK may not be worth the computational effort required? BQSR seems to be the polar opposite of binning. Was the BQSR step included in the GATK pipeline used in the analysis?
John Farrell
March 7, 2013 at 4:10 pm
John;
I’d like to investigate the influence of BQSR, but these results don’t give you a definite answer to that question. BQSR is included in the variant calling pipeline, and I tested binning and not-binning after BQSR. Both of these give similar concordance numbers which suggests that the post-BQSR precision does not need 40-quality values. However, it doesn’t answer how leaving out the BQSR step entirely influences the final variant calls.
I’m currently working on evaluation tools to help investigate this, and also the impact of changing aligners. Since I expect there will be trade offs, it’ll be useful to be able to assess characteristics (low coverage, repetitive, mappability, indel size…) of changed variant calls.
Brad Chapman
March 7, 2013 at 8:09 pm
[…] always suspected that BQSR is of little benefit, but I stand corrected here. So, similar to Brad Chapman’s results, this seems to indicate that base quality rebinning does not reduce the quality of variant calls. […]
Base quality score rebinning | BaseCall
April 23, 2013 at 8:31 am